Las canalopatías cardíacas son un grupo de síndromes que afectan el sistema eléctrico del corazón. En la mayoría de los pacientes con canalopatías, la estructura del corazón es normal. En un corazón normal y saludable, las cámaras del corazón se contraen rítmicamente, bombeando sangre a través del cuerpo. Señales eléctricas que se originan en el sistema eléctrico causan que las células musculares del corazón (miocitos) se contraigan. Estas señales eléctricas dependen del flujo de sodio, potasio y iones de calcio dentro y fuera de las celdas. Los iones pasan a través de canales (a veces llamado “canales de proteína”) en las membranas celulares para crear la señal eléctrica que genera cada latido del corazón
Si los canales del ion no funcionan correctamente, la estabilidad eléctrica del corazón y el ritmo cardíaco pueden volverse anormales. Ritmos cardíacos anormales (arritmias) puede provocar desmayos o paros cardíacos y muerte súbita. Un paro cardíaco no es lo mismo que un ataque al corazón, aunque ambos pueden ser fatales. Un paro cardíaco es una interrupción repentina del corazón por razones eléctricas. Un ataque al corazón es la falta de flujo sanguíneo al músculo cardíaco, generalmente debido a una enfermedad en las arterias.
La causa de la función anormal de los canales iónicos generalmente se hereda. Esto significa que uno de los genes que codifican las proteínas no es normal. Si este gen se pasa de padres a hijos, se forma un canal de iones anormal. En raras ocasiones, una mutación puede ocurrir espontáneamente. En estos casos, no habrá antecedentes familiares de la condición.
La mayoría de las muertes debidas a canalopatías hereditarias se pueden prevenir si se diagnostican las condiciones. El diagnóstico de una canalopatía cardíaca debe intuirse en cualquier persona joven con síncope inexplicado (pérdida repentina de la conciencia o desmayo), paro cardíaco o muerte súbita. El síncope es el síntoma principal y puede ser el único signo de advertencia de canalopatías cardíacas hereditarias. No todas las personas afectadas mostrarán síntomas. La falta de síntomas previos, por lo tanto, no significa necesariamente que un paciente o una familia no tengan una canalopatía hereditaria. Sin embargo, muchas personas que mueren repentinamente de canalopatías tenían síntomas previos que fueron ignorados, diagnosticados erróneamente o considerados insignificantes. Si tiene alguno de los síntomas, es muy importante que los informe a su médico de cabecera.
Se debe considerar un diagnóstico de canalopatía cardíaca en cualquiera de las siguientes situaciones:
• Una persona joven con síncope o convulsiones inexplicables, especialmente en situaciones de ejercicio o estrés emocional.
• Un sobreviviente de un paro cardíaco inexplicado.
• Antecedentes familiares de síncope inexplicado, convulsiones o muerte súbita. Se considera que una muerte es “inexplicable” cuando una autopsia no encuentra una causa evidente de muerte.
Si tiene algún síntoma sospechoso, siempre es mejor buscar atención médica para una evaluación básica. Esto es especialmente cierto cuando se sospecha que un desmayo es derivado de una arritmia. Estos desmayos “de advertencia” son generalmente repentinos e inesperados, que ocurren durante el ejercicio físico o el estrés emocional. Por el contrario, un desmayo común generalmente tiene una causa bastante obvia, como dolor, lesión o una experiencia desagradable, y está precedida por síntomas de advertencia como mareos, visión borrosa, hormigueo, náuseas o sudoración.

Cuando un individuo ha sido diagnosticado con una arritmia hereditaria, todos los familiares principales (padres, hermanos, hijos) también deben ser evaluados. Deben someterse a pruebas de rutina, como pruebas de ECG, Holter y cinta rodante. Si el miembro de la familia afectado ha sido identificado con una mutación específica, también considere realizar pruebas genéticas.
Una vez identificadas, estas enfermedades a menudo se pueden manejar muy bien. En su mayor parte, la muerte súbita se puede prevenir y puede continuar viviendo una vida plena.
Hay muchos tipos diferentes de mutaciones genéticas que finalmente conducen a una variedad de canalopatías cardíacas. Estas canalopatías pueden dar lugar a una serie de síndromes, incluidos:
Síndrome de Brugada (BrS)
Taquicardia ventricular polimórfica catecolaminérgica (CPVT)
Síndrome de QT corto (SQTS)
En la mayoría de los casos de estas afecciones, los tratamientos farmacológicos preventivos simples controlan la mayoría de los síntomas. Sin embargo, algunas personas o familias se benefician al mantener un desfibrilador externo automático (DEA) en el hogar. Consulte esta sugerencia con su médico. Las siguientes secciones brindan información más detallada sobre cada una de las cuatro canalopatías más comunes.
El síndrome de Brugada (BrS) es una canalopatía hereditaria que ocasiona anomalías en el estado eléctrico del músculo cardíaco. El sello distintivo de esta condición es el hallazgo ECG característico que refleja la anomalía eléctrica. Las personas con BrS tienen riesgo de arritmias que pueden provocar síncope o muerte súbita. BrS afecta a más hombres que mujeres. La edad promedio a la que aparecen los síntomas es de 40 años; aunque se han reportado síntomas en pacientes con edades desde recién nacidos hasta los 84 años.
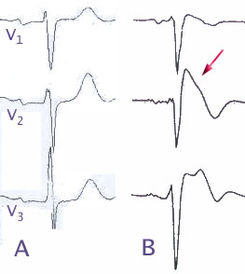
El BrS es una enfermedad hereditaria que resulta de mutaciones en uno o más de varios genes del canal iónico diferentes. Los genes que llevan instrucciones para los canales de sodio y calcio en los músculos del corazón se afectan con mayor frecuencia. Aunque se han identificado algunos defectos genéticos específicos que conducen al BrS, es probable que haya otras mutaciones que aún no se hayan descubierto.
El síncope y el paro cardíaco son las indicaciones más comunes que conducen al diagnóstico de BrS. El paro cardíaco se produce con mayor frecuencia durante el sueño o el descanso, más que durante la actividad física. Puede haber antecedentes de problemas para respirar por la noche. La fiebre y los medicamentos parecen desencadenar o empeorar los síntomas del síndrome de Brugada en muchos. Algunos pacientes, sin embargo, no muestran síntomas obvios. Puede haber un antecedente familiar de muerte cardíaca repentina, aunque no universal, ya que el síndrome puede ocurrir en un individuo sin antecedentes familiares conocidos con la enfermedad.
Su médico puede suponer que tiene BrS en función de sus síntomasy / o antecedentes familiares. Los patrones característicos en un electrocardiograma (ECG) también pueden sugerir el diagnóstico o utilizarse para confirmarlo. Estos patrones pueden estar presentes todo el tiempo, pueden aparecer y luego normalizarse espontáneamente, o pueden ser provocados por la fiebre o la administración de determinados medicamentos. El BrS se puede descubrir solo cuando un electrocardiograma de rutina (ECG) muestra un patrón anormal conocido como elevación del segmento ST. Muchas personas tienen otras anomalías del ECG, como bloqueo cardíaco o fibrilación auricular.
El BrS a veces es difícil de diagnosticar. Muchas personas que tienen un gen BrS pueden nunca experimentar ningún síntoma de advertencia. El mal funcionamiento eléctrico puede o no aparecer en un ECG.
El rendimiento de las pruebas genéticas sigue siendo relativamente bajo: solo del 11 al 28% de las personas con un diagnóstico de BrS tienen un gen BrS identificado. Las pruebas genéticas pueden ayudar en la identificación temprana de los miembros de la familia en riesgo potencial. Si ha tenido un diagnóstico clínico positivo de BrS, sus hijos, hermanos y padres deben ser evaluados.
Consulte el uso de estos medicamentos potencialmente peligrosos con su cardiólogo. Adicionalmente, la fiebre puede provocar cambios eléctricos en el corazón en el BrS, y la fiebre debe tratarse de forma agresiva con antipiréticos (acetaminofén o ibuprofeno). Si la fiebre no responde a los medicamentos antipiréticos, el monitorización cardíaca es importante hasta que la fiebre se resuelva.
Debido a que el ECG de Brugada se puede malinterpretar fácilmente como un “ataque cardíaco”, es importante que lleve consigo una copia de su ECG cada vez que busque atención médica e informe a los profesionales de la salud de su condición.
Su médico puede suponer que tiene BrS en función de sus síntomasy / o antecedentes familiares. Los patrones característicos en un electrocardiograma (ECG) también pueden sugerir el diagnóstico o utilizarse para confirmarlo. Estos patrones pueden estar presentes todo el tiempo, pueden aparecer y luego normalizarse espontáneamente, o pueden ser provocados por la fiebre o la administración de determinados medicamentos. El BrS se puede descubrir solo cuando un electrocardiograma de rutina (ECG) muestra un patrón anormal conocido como elevación del segmento ST. Muchas personas tienen otras anomalías del ECG, como bloqueo cardíaco o fibrilación auricular.
El BrS a veces es difícil de diagnosticar. Muchas personas que tienen un gen BrS pueden nunca experimentar ningún síntoma de advertencia. El mal funcionamiento eléctrico puede o no aparecer en un ECG.
No todos los médicos profesionales están de acuerdo en si los pacientes con BrS deben participar en actividades físicas de alta intensidad. Debería discutir este problema con su cardiólogo
La taquicardia ventricular polimórfica catecolaminérgica (TVPC) se caracteriza por ritmos cardíacos anormales (arritmias) que pueden provocar síncope o paro cardíaco. Estos eventos usualmente ocurren durante el ejercicio físico o con estrés emocional. La edad promedio a la que aparecen los síntomas de CPVT es entre siete y nueve años, aunque los síntomas pueden aparecer hasta la cuarta década de vida. Si no se trata, esta condición puede ser letal. Hasta 80% de las personas diagnosticadas experimentan uno o más episodios sincopales y aproximadamente un 30% experimentan paro cardíaco. Se piensa que el CPVT es mucho menos común que otras canalopatías, aunque afecta a bebés, niños, adolescentes y adultos sanos. La verdadera prevalencia de CPVT sigue siendo desconocida, pero se estima que es de aproximadamente 1 en 10,000. Con mayor conocimiento, pruebas genéticas y opciones de tratamiento efectivas, los médicos ahora pueden diagnosticar CPVT temprano y proporcionar tratamiento para prevenir la muerte súbita.
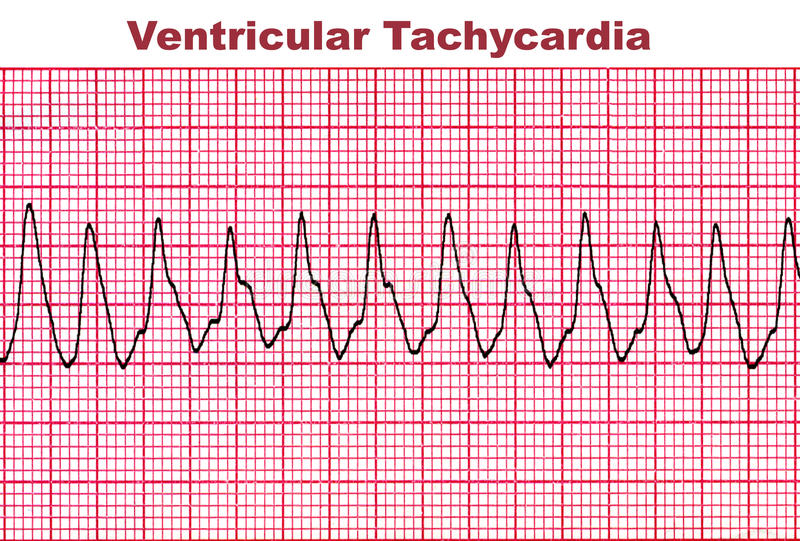
CPVT es una canalopatía hereditaria que resulta de mutaciones en uno o más de los genes que manejan el calcio dentro de las células del músculo cardíaco. Las arritmias pueden ser desencadenadas por condiciones que involucran estrés emocional o físico que lleva a una liberación de adrenalina.
Los pacientes con CPVT pueden experimentar palpitaciones, convulsiones o síncope cuando el cuerpo produce altos niveles de adrenalina, como durante el ejercicio o en momentos de estrés. Se supone CPVT cuando un individuo tiene un episodio de síncope, convulsiones o paro cardíaco asociado con ejercicio o estrés emocional. También se supone CPVT cuando la autopsia es normal después de la muerte súbita e inesperada de una persona joven.
Las pruebas de CPVT incluyen un ECG en reposo (que generalmente es normal) y una prueba de esfuerzo (caminar en una cinta de correr o andar en bicicleta mientras se registra el ritmo cardíaco con un ECG) para tratar de provocar las anomalías del ritmo. Niños que son demasiado jóvenes para una cinta de correr o una prueba de bicicleta pueden ser monitoreados durante las actividades rutinarias usando un monitor Holter. Una alternativa para las pruebas de esfuerzo en cinta rodante en alguien que no puede hacer ejercicio es la prueba de provocación con catecolaminas, en la cual se administra adrenalina por vía intravenosa mientras se controla el ritmo cardíaco. Se realiza un diagnóstico de CPVT cuando se observan las arritmias características. Estos incluyen latidos ventriculares prematuros frecuentes y arritmias ventriculares, como taquicardia ventricular bidireccional, que pueden provocar fibrilación ventricular.
Hay dos genes conocidos que causan CPVT. Ambos afectan a hombres y mujeres por igual. El gen de CPVT más común es dominante, mientras que una forma rara es recesiva. Una vez que un miembro de la familia se identifica con CPVT, otros familiares cercanos deben buscar pruebas para el síndrome.
No hay cura para CPVT, pero hay tratamientos efectivos. Todos los pacientes que tienen síntomas de CPVT deben recibir tratamiento. Todos los niños y adultos jóvenes con CPVT deben recibir tratamiento incluso si no tienen síntomas. Esto se debe a que los síntomas pueden aparecer en cualquier momento, siendo la muerte súbita el primer síntoma.
El tratamiento habitual consiste en tomar un medicamento llamado betabloquedor todos los días. Los betabloqueadores previenen la respuesta del corazón a la adrenalina, reduciendo la probabilidad de arritmia. Las pruebas de esfuerzo repetidas en cinta rodante pueden ayudar a determinar la dosis correcta. Si el betabloqueante no controla adecuadamente la arritmia, también se ha encontrado que un medicamento adicional, Flecainida, es útil. Estos medicamentos son usualmente muy exitosos para controlar las arritmias si se toman con regularidad. Necesitará un seguimiento regular con su médico para asegurarse de que su arritmia esté bien controlada. Los niños en crecimiento necesitan que su dosis de medicamento se ajuste regularmente. Debe informar cualquier síntoma recurrente.
Si continúa teniendo síntomas a pesar de las dosis adecuadas de medicamentos, hay otros tratamientos. Un tratamiento es la denervación simpática cardíaca (LCSD). Esta es una cirugía en la que se eliminan los nervios que son responsables del suministro de adrenalina al corazón. En raras ocasiones, se necesita un ICD.
Es muy importante que tome sus medicamentos todos los días. Los padres deberían enseñar a sus niños sobre la importancia de la medicación diaria, y deberían asegúrese de tomar cada dosis del día. Los preadolescentes y adolescentes también deben ser supervisados cuando toman sus medicamentos. Debido a que el ejercicio físico extenuante aumenta el riesgo de arritmia, debe evitar actividades competitivas o de alta intensidad. Su cardiólogo consultará esas actividades con un menor riesgo de arritmia.
También se le pedirá que evite el uso de medicamentos que puedan estimular el corazón, como medicamentos para el resfriado de venta libre o algunos inhaladores para el asma.
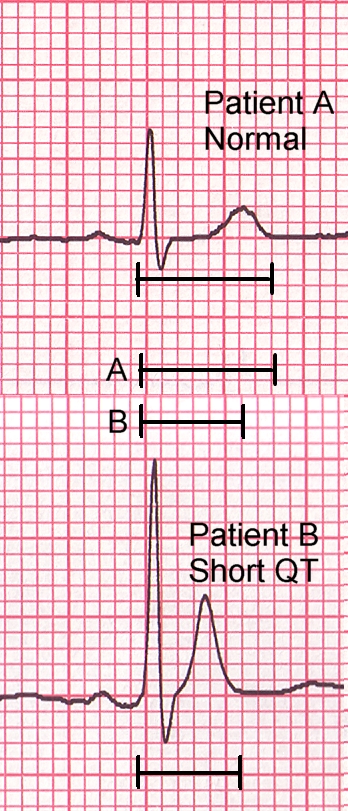
El síndrome de QT corto (SQTS) es una rara afección cardíaca que con lleva el riesgo de arritmias, que pueden provocar síncope o muerte súbita. En esta condición, la recuperación eléctrica del músculo cardíaco después de cada latido del corazón es anormal. Esto es evidente en el ECG como acortamiento del intervalo QT (Figura 9). El SQTS parece ser raro, pero la condición puede estar infradiagnosticada, pues algunas personas afectadas nunca experimentan síntomas.
El SQTS es una canalopatía hereditaria que resulta de una mutaciones en uno o más de los genes que manejan el movimiento de iones dentro y fuera de las células del músculo cardíaco.
El síncope es el síntoma más común del SQTS. Otros síntomas pueden ser convulsiones o incluso muerte súbita. Muchas personas también tienen otras anomalías del ECG, como bloqueo cardíaco o fibrilación auricular. Muchas personas con SQTS se dan cuenta de su afección solo a partir de los resultados de un ECG realizado por un motivo no relacionado, ya que tienen antecedentes familiares con SQTS, o debido a los resultados de pruebas genéticas.
Las pruebas de SQTS incluyen un ECG de reposo (que puede ser normal) y una prueba de esfuerzo: caminar en una cinta de andar o andar en bicicleta mientras se registra el ritmo cardíaco con un ECG.
Estas pruebas le permiten a su cardiólogo observar el comportamiento eléctrico del corazón con diferentes ritmos cardíacos.
Seis genes están asociados con el SQTS. Se puede identificar una mutación genética en aproximadamente el 25% de las personas con un diagnóstico clínico de SQTS. En los casos en que los resultados de las pruebas genéticas para mutaciones conocidas sean negativos, la detección familiar solo se puede llevar a cabo con un ECG y pruebas de estrés.
No hay cura para el SQTS, pero hay tratamientos efectivos. Consulte las opciones de tratamiento con su cardiólogo. Todos los pacientes que tienen síntomas deben recibir tratamiento. En la mayoría de los casos, los médicos recomiendan que todos los niños y adultos jóvenes con SQTS, incluso aquellos sin síntomas, deben de ser tratados. Los síntomas pueden ocurrir en cualquier momento, siendo la muerte súbita, a veces el primer síntoma.
La colocación de un DAI es un tratamiento efectivo de primera línea para prevenir la muerte súbita cardíaca. Para el tratamiento de la arritmia, existen medicamentos que se pueden usar para prolongar el intervalo QT, lo que reduce efectivamente el riesgo de muerte súbita. Estos medicamentos incluyen quinidina, que está disponible en Canadá si su médico hace una solicitud especial para usarla en su caso.
No todos los profesionales médicos están de acuerdo en si los pacientes con SQTS deben participar en actividades físicas de alta intensidad. Debería consultar este problema con su cardiólogo.
Adrenalina: una hormona producida por el cuerpo que tiene el efecto de acelerar el corazón; también llamado epinefrina.
Arritmia: un ritmo cardíaco irregular potencialmente letal.
Fibrilación auricular: contracción irregular de las aurículas del corazón.
Desfibrilador externo automático (DEA): dispositivo que detecta un ritmo cardíaco anormal (fibrilación) y envía una descarga eléctrica para detener la fibrilación y permitir que el corazón retome el ritmo normal.
Betabloqueante: medicamento que trata los ritmos cardíacos anormales al bloquear la respuesta del corazón a la adrenalina.
Paro cardíaco: una parada repentina del corazón potencialmente fatal, debido a un mal funcionamiento del sistema eléctrico del corazón.
Prueba de provocación con catecolaminas: una técnica de diagnóstico para provocar malos ritmos cardíacos que de otra manera no podrían expresarse; Administración IV de epinefrina (adrenalina) mientras un paciente se somete a un ECG para controlar la respuesta del corazón al medicamento
Electrocadiograma (ECG): gráfico que muestra la actividad eléctrica del corazón a lo largo del tiempo.
Estudio de electrofisiología (EPS): un examen médico para investigar las anomalías del sistema eléctrico del corazón; implica la colocación de electrodos en el corazón.
Pruebas genéticas: análisis del material genético de un individuo con el fin de identificar ciertos genes.
Monitor de Holter: técnica de diagnóstico en la cual la actividad eléctrica del corazón de un paciente se mide con un dispositivo portátil durante un período prolongado.
Desfibrilador cardioversor implantable (DAI): dispositivo implantado en el pecho que detecta cualquier arritmia en el corazón y administra una descarga eléctrica para detener la fibrilación y permitir que se reanude la función cardíaca normal.
Ion: una partícula cargada de un elemento, por ejemplo, sodio, Na +; calcio, Ca2 +
Canal de iones: una proteína en la membrana celular que mueve los iones dentro o fuera de la célula, manteniendo así una diferencia de potencial eléctrica a través de la membrana celular.
Mutación: un cambio en la secuencia de material genético que a menudo afecta la estructura de las proteínas en el cuerpo.
Miocitos: células musculares dentro del corazón.
Palpitaciones: la sensación de un latido del corazón inusualmente rápido.
Elevación del segmento ST: una característica de un electrocardiograma en el que una porción del trazado es inusualmente alta.
Prueba de esfuerzo: una prueba de diagnóstico en la que el paciente realiza actividad física (a menudo en una cinta de correr) mientras se somete a una electrocardiografía para registrar la actividad eléctrica del corazón.
Muerte cardíaca súbita: muerte inesperada como resultado de la pérdida de la función cardíaca.
Síncope: pérdida repentina de la conciencia sin causa aparente.
Síndrome: una colección de síntomas con una causa relacionada.
Fibrilación ventricular: una contracción desorganizada de los músculos del corazón que no produce una contracción adecuada del ventrículo, por lo que la sangre ya no se bombea alrededor del cuerpo.
Taquicardia ventricular: contracción rápida de los ventrículos del corazón.